سندروم آشر یک بیماری ژنتیکی است که هم بر شنوایی و هم بر بینایی فرد تاثیر می گذارد. از این سندروم به عنوان یک بیماری و یا اختلال میتوان نام برد که بیش از یک علامت و ویژگی دارد. بزرگترین علامت این سندروم از دست دادن شنوایی و اختلال در بینایی است که این اختلال می تواند منجر به عارضه از دست دادن بینایی در شب ( شب کوری) شود. به طور کلی سه تا شش درصد کودکانی که ناشنوا هستند و سه تا شش درصد کودکانی که کم شنوا هستند به دلیل این سندروم دچار این اختلالات شنوایی شده اند. سندروم آشر یک عامل وراثتی است و این موضوع به این معنی ست که در صورت ابتلای والدین به این سندروم احتمال ابتلای کودکان به این سندروم نیز وجود دارد، این انتقال از طریق ژن ها از والدین به کودک صورت می گیرد. عامل وراثت در سندروم آشر به این صورت عمل می کند که فرد باید هم از پدر و هم از مادر این ژن وراثتی را دریافت کند و در صورتی که فقط از یکی از والدین این ژن دریافت شود، ابتلا به این سندروم امکان پذیر نخواهد بود. در صورتی که این ژن هم در پدر و هم در مادر وجود داشته باشد و احتمال اینکه فرزند آنها دچار سندروم آشر شوند یک به چهار است. باید گفت به طور کلی سه نوع سندروم آشر وجود دارد که در هر سه نوع آنها عامل ژن های وراثتی نقش اصلی را ایفا می کنند، این دسته بندی به شرح زیر است: نوع اول این سندروم کودکانی هستند که با ناشنوایی با درجه ی شدیدی به دنیا می آیند که وجود مشکلات تعادلی در این کودکان به وفور دیده می شود. کودکانی که با نوع دوم از سندروم آشر متولد می شوند دارای مشکلات شنوایی هستند و در حفظ تعادل نیز دچار مشکل اند اما لازم به ذکر است که مشکل حفظ تعادل در این گروه خیلی کمتر از کودکان مبتلا به سندروم آشر در گروه اول است. نوع سوم از کودکانی که دچار سندرم آشر هستند در هنگام تولد شنوایی سالمی داشته و به صورت کلی این کودکان تقریبا همانند افراد عادی می توانند تعادل خود را حفظ نمایند و مشکلات تعادلی در آنها به مرور زمان اتفاق می افتد. مشکلات شنوایی در این نوع افراد به مرور زمان و با بالا رفتن سن این کودکان غالبا در سنین نوجوانی پدیدار می شود و این افراد مجبور به استفاده از سمعک های شنوایی در اواسط دوره ی بزرگسالی خود هستند. از آن جایی که سندروم آشر اختلالی در شنوایی، بینایی و تعادل فرد ایجاد می کند در نتیجه برای تشخیص آن انجام تست های بینایی و انواع تست های شنوایی در اسرع وقت توصیه می شود. همچنین در آزمایشگاه ژنتیک نسل سالم می توان این بیماری را با انجام تست های ژنتیکی تشخیص داد و از انتقال آن به نسل بعد جلوگیری کرد.
خلق خو شامل صفات رفتاری نظیر معاشرت پذیری، احساسی بودن، سطح فعالیت (انرژی بالا یا پایین)، سطح توجه، و مصر بودن میباشد. خلق و خو به ویژه در سرتاسر بزرگسالی نسبتا ثابت باقی میماند. خلق و خوی مشابه در یک خانواده به ژنتیک یکسان و به محیطی که فرد در آن بزرگ شده است نسبت داده میشود. مطالعات دوقلوهای همسان (کسانی که صد درصد DNA یکسان دارند) نشان میدهد که ژنتیک نقش بزرگی در این قضیه ایفا میکند. دوقلوهای همسان اغلب در مقایسه با سایر خواهر برادرهایشان خلق و خوی یکسانی دارند. حتی دوقلوهای همسانی که دور از یکدیگر در خانوادههای جدا بزرگ شده باشند نیز در این صفات مشترک هستند. دانشمندان برآورد میکنند که بیست تا شصت درصد از خلق و خو توسط ژنتیک تعیین میشود. اما خلق و خو الگوی وراثت روشنی ندارد و ژنهای خاصی وجود ندارد که صفت رفتاری خاصی را به ما بدهد. در عوض بسیاری از اختلافات ژنی مشترک (پلی مورفیسم ها) برای اثر گذاری بر خلق و خوی یک فرد با هم همراه میشوند. سایر تغییرات DNA که توالیها را تغییر نمی دهند (تغییرات اپی ژنتیک) نیز در خلق و خو دست دارند. مطالعات زیادی ژنهای متعددی را شناسایی کرده اند که در خلق و خو نقش دارند. بسیاری از این ژنها در ارتباطات بین سلولها و مغز دست دارند. تغییرات ژنی خاصی در صفات خاص که به خلق و خو مربوط میشوند نقش دارند. برای مثال تغییرات ژن های DRD2 و DRD4 با تمایل به جستجوی تجارب جدید، و ژن KATNAL2 با منظم بودن و با احتیاط بودن ربط داده شده اند. تغییراتی که روی ژنهای PCDH15 , WSCD2 اثر میگذارند با معاشرت پذیری ارتباط دارند، درحالیکه برخی اختلافات ژنی MAOA با درون گرایی، به ویژه در محیطهای خاص ارتباط دارند. تغییرات ژنهایی نظیر SLC6A4, AGBL2 در اضطراب و افسردگی نقش دارند. فاکتورهای محیطی با اثر گذاری بر فعالیت ژن ها نیز در خلق و خو دخیلند. در کودکانی که در یک محیط نامناسب بزرگ میشوند، ژنهای افزایش دهندهی صفات رفتاری بی ملاحظگی و بی پروایی روشن (فعال) میشود. اما کودکی که در محیط مثبت رشد میکند خلق و خوی آرام تری دارد، که تا حدی به دلیل ژنهای متفاوتی است که روشن میشود.
آنمی فانکونی یک بیماری ژنتیکی است که در نتیجه نقص در شاخهای از پروتئینهای مسئول در ترمیم DNA ایجاد میشود. در نتیجه در بسیاری از بیماران سرطان شایعترین آن اغلب لوسمی میلوژنوس حاد و در نود درصد موارد نارسایی مغز استخوان رخ میدهد. این بیماری اصولاً بر مغز استخوان تأثیر می گذارد. بیماران مبتلا به کم خونی فانکونی دارای تعداد سلول سفید، سلول قرمز خون و پلاکت ( سلولهایی که به انعقاد خون کمک می کنند) کمتری نسبت به میزان طبیعی هستند. سلولهای سفید ناکافی خون می توانند منجر به عفونت شوند. فقدان سلولهای قرمز خون ممکن است منجر به خستگی ( کم خونی) شود. میزان پایین تر از حد طبیعی پلاکت ممکن است منجر به خونریزی شدید گردد. در ۱۰ درصد موارد این بیماران دچار عقب ماندگی ذهنی هستند. اکثر افراد مبتلا به کم خونی فاکونی این نوع علائم را دارند: قلب، شش ها و مجرای گوارشی غیر طبیعی، مشکلات استخوان (به ویژه مفاصل ران، ستون فقرات یا دنده ها) می توانند باعث انحنای ستون فقرات (اسکولیوز) شوند. تغییراتی در رنگ پوست، وجود نواحی تیره بر روی پوست، بیماری پوستی با ایجاد لکه های روشن بر روی پوست (Vitiligo)، ناشنوایی در رابطه با غیر طبیعی شدن گوش، مشکلات چشم یا پلک، کلیه (هایی) که بدرستی شکل نگرفته اند، مشکلاتی همراه با بازوها و دست ها مثل: از دست دادن انگشت شست، وجود انگشت شست اضافی یا بد شکل، مشکلات دست ها و استخو ان در پایین بازو، وجود استخوان کوچک یا از دست دادن استخوان در ساعد، کوتاه قدی، سر کوچک، بیضه های کوچک و تغییرات تناسلی. برای به ارث بردن ایننوع کمخونی فرد می بایست یک کپی از ژن غیر طبیعی از هر والد گرفته باشد. تشخیص ژنتیکی این بیماری و جلوگیری از انتقال آن به نسل بعد در آزمایشگاه ژنتیک نسل سالم انجام می شود.
تصور کنید برای یک بیماری ساده یا پیچیده به پزشکتان مراجعه
کردهاید. پزشک نمونهای از خون یا موی شما را میگیرد و با تحلیل ترکیب ژنتیکی
شما یک داروی کاملا اختصاصی برایتان تجویز میکند.
گرچه این مساله در حال حاضر به صورت عملی برای درمان بیشتر
بیماریها امکان پذیر نیست، اما آینده علم دارو و درمان به سمت تولید داروهای
اختصاصی برای هر فرد و براساس اطلاعات ژنی وی پیش میرود.
مطالعات دارویی و درمانی و اثرات متقابل دارو بر متابولیسم
افراد سبب شده تا محققان به این نتیجه برسند که بهترین روش درمان تولید و تجویز
داروهای اختصاصی براساس ویژگیهای ژنتیکی افراد است. این مساله میتواند اثربخشی
دارو را افزایش دهد و از عوارض جانبی آن بکاهد.
در همین راستا مطالعات فارماکوژنومیکس به کمک دانشمندان میآیند.
در فارماکوژنومیکس تفاوتهای ژنتیکی موروثی در مسیرهای متابولیسم داروهایی بررسی
میشوند و به بررسی نقش تفاوتهای ژنتیکی در ارتباط با واکنش به داروها از طریق
بررسی سیستماتیک ژنها، محصولات ژنی و تغییرات فردی در بیان و عملکرد ژن میپردازد.
در صورت عملی شدن این مساله، افرادی که بیماری آنها به طور
مستقیم با ژنتیکشان مرتبط است سود بسیاری میبرند. به این ترتیب شانس درمان بیماریهای
روانی مانند افسردگی، انواع سرطانها، بیماریهای مغزی مانند زوال مغز و
نورودژنراتیوهمچون آلزایمر و پارکینسون، که با ژنتیک افراد رابطه مستقیمی دارند،
افزایش قابل توجهی خواهد یافت.
تاریخ فارماکوژنومیکس به ۵۱۰ سال پیش از میلاد
مسیح باز میگردد؛ زمانی که فیثاغورس متوجه شد که استفاده از باقلا سبب مرگ برخی
افراد خواهد شد اما بر برخی دیگر بیاثر است. با این وجود مطالعات جدید در این
زمینه از نیمه دوم قرن بیستم میلادی و به دنبال افزایش میزان دانش بشر در مورد علم
ژنتیک آغاز شد.
فارماکوژنتیکس و فارماکوژنومیکس به موضوعاتی بحث برانگیز در
زمینه اخلاق زیستی و پزشکی تبدیل شدهاند. فراگیر شدن این علوم که درمان بیماریهای
رایج و نادر را شامل خواهد شد تاثیر زیادی بر جامعه خواهد گذاشت.
به دلیل نوپا بودن این علوم سوالات زیادی در رابطه با مسائل
اخلاقی و راهحلهای احتمالی مقابله با مشکلات انسانی مطرح شده است. به عنوان مثال
دسترسی به اطلاعات ژنتیکی افراد تا چه اندازه ممکن است در زندگی خصوصی آنها اهمیت
داشته و حریم خصوصی آنها را به خطر بیاندازد. به علاوه مسائلی چون ایمنی دارو،
نحوه دسترسی به آن و برقراری عدالت در درمان بیماران نیز از مسائلی است که باید
مورد توجه قرار گیرد.
دیربازی است که بحث
طراحی داروهای منحصر به فرد برای هر بیمار مطرح است اما به نظر می رسد این موضوع
شتاب جدی در عرصه دارویی پیدا کرده و درصورتیکه برنامه ریزی برای مواجه با این
حوزه نوآورانه در سیاست های وزارت بهداشت و سازمان دارو غذا منظور نگردد دیر یا زود با مشکلاتی در مدیریت
بازار داروهایی که بر مبنای این ویژگی به بازار دارویی وارد می شوند مواجه می گردیم
ایجاد زیرساخت لازم و ظرفیت آزمایشگاهی برای ارزیابی ژنتیکی بیماران و هدایت آنها
به سمت مصرف و یا عدم مصرف یک دارو از پیش نیازهای تسلط بر این بازار خواهد بود.
کشف دارو و توسعه دارو
بیوانفورماتیک و نقش
فارماکوژنومیکس در فرآیند کشف دارو و فرآیند توسعه بیوانفورماتیک
بیوانفورماتیک و نقش
فارماکوژنومیکس در فرآیند کشف و توسعه دارو
بیوانفورماتیک و نقش
فارماکوژنومیکس در فرآیند کشف دارو و فرآیند توسعه
فارماکوژنومیکس و بیوانفورماتیک
به اثرات پلی مورفیسم ژنتیکی و انواع ژنومی در واکنش دارو اشاره میکند، دانش آن میتواند
به انتخاب فرآیند بهینه دارو، دوز و درمان کمک کند و از واکنش های منفی دارو
اجتناب کند. دانش زیستی ژنتیکی و بیوانفورماتیک مرتبط با بیماری، شرکتهای داروسازی
را به طراحی فارماکوژنومیکس و دوزهای فردی و دوزهای فردی هدایت میکند. این کار با
مشاهده الگوهای ژنتیکی و پلی مورفیسم این عناصر ژنتیکی انجام میشود که تعامل با
دارو یا محصولات فرعی آن را نشان میدهد و به طریقی با فارماکوژنومیکس دارو مرتبط
میشود. عوارض جانبی داروها به عنوان یکی از دلایل اصلی مرگ در بین بیماران بستری
گزارش شدهاست و اکثریت ۱۷
تا ۲۹ میلیارد دلار هزینههای
سالانه اشتباه ات پزشکی را تشکیل می دهد. واکنشهای جانبی دارویی همچنین به عنوان
عامل از دست دادن اعتماد در سیستم بهداشت و درمان و رضایت رو به کاهش هم بیماران و
هم متخصصان بهداشتی گزارش شده است.
دارو و پزشکی شخصی
رویکرد فارماکوژنومیکس
، شرکتهای داروسازی را قادر میسازد تا داروهایی را طراحی کنند که الزامات زیر
گروههای ژنتیکی خاص جمعیت عمومی را برآورده سازند. هدف اصلی فارماکوژنومیکس و بیوانفورماتیک
شناسایی بیمارانی است که اثر بخشی دارو را می توان پیش بینی کرد و به منظور کاهش
ریسک اثرات مضر دارو از آن افراد استفاده کرد .وعده ی تجویز داروها براساس پروفایل
ژنتیکی بیماران به عنوان “داروی شخصی” شناخته میشود. این کار باعث کاهش گمانه زنیهای
مصرف داروهای تجویزی ساتفاده میشود و در نتیجه اعتماد به پزشک و هم بیمار را افزایش
میدهد و روشهای غالب برای کشف دارو و توسعه ، تشخیص، درمان و استراتژیهای پیشگیری
از بیماریها را اصلاح میکند. همچنین برای جامعه نیز مفید است چون مصرف داروهای
گران قیمت در بیمارانی که اختلالات آن توسط این داروها درمان نمیشوند، اجتناب میشود.
بیوانفورماتیک همچنین منابع اطلاعاتی مربوط به فارماکوژنومیکس را فراهم میکند که
حاوی اطلاعاتی در مورد انواع مختلف پلی مورفیسم بوده و واکنش متغیر وابسته به دارو
را بررسی می کند گزارش شده است که دارو های مختلف ، واکنشهای منفی دارو را نشان
میدهند که اغلب منجر به بستری شدن و در برخی موارد موجب تلفات میشود. تحقیقات
درباره چنین واکنشی به دارو منجر به خروج دارو از بازار شده است. این مساله
بلافاصله توسط مجموعهای از پروندههای حقوقی برای سو درمان دارویی پی گیری میشود.رویکرد
فارماکوژنومیکس برای استراتژی توسعه دارو فرصتی برای معکوس کردن این روند است.
وعده داده شده است که ممکن است منجر به “توسعه دقیق مواد دارویی” شود. داروهای با
دقت به داروهایی اشاره میکنند که با ترکیب ژنتیکی افراد متناسب هستند. این داروها
را می توان در آزمایشهای کلینیکی کوتاه و ساده ارزیابی کرد و اثرات نامطلوبی بر
روی آنها نشان خواهد داد را ارزیابی کرد.تایید و آزمایش ژنتیکی قبل از تجویز دارو
و آنالیز بیوانفورماتیکی به شدت شانس نسخه غلط دارو را کاهش میدهد.
استفاده از دارو های بی مصرف
در فرآیند کشف دارو و
توسعه دارو ، شرکتهای داروسازی بیشتر بر روی داروهای اصلی متمرکز میمانند که به
بیش از ۲۰ میلیون نفر توصیه میشود
، این داروها به عنوان داروهای Blockbuster شناخته میشوند. نتیجه این کار فقدان مواد دارویی است که ممکن
است برای درمان بیماریهایی که تنها بر تعداد کمی از مردم تاثیر می گذارند، توسعه
داده شود.این داروها – یا داروهای بالقوه به عنوان دارو های
رها شده یا یتیم شناخته میشوند. یک استراتژی فارماکولوژی برای توسعه دارو ممکن
است این داروهای رها شده را احیا کند اگر بتوان ثابت کرد که ذینفعان بالقوه برای این
داروها وجود دارند . از نقطه نظر تجاری، اگر شرکت داروسازی بتواند از چیزی مانند
وضعیت دارو های رها شده در محصول خود بهره مند شود، این کار به تشویق طبقه بندی
جمعیت ها براساس فارماکوژنتیکس با بیوانفورماتیک کمک خواهد کرد، زیرا کاهش اندازه
یک جمعیت میتواند با اولویت دارویی جبران شود. این تنها راهی است که شرکتهای
داروسازی میتوانند ترغیب شوند تا این تعصب
blockbuster را رها کنند. در سال
های اخیر، مقامات مختلف منابع و طراحی دارو های با پتانسیل فارماکوژنومیکس را تشخیص
دادهاند و چنین روشهایی را برای کشف دارو و فرآیند تحویل تشویق کردهاند.همانطور
که فن آوریهای فارماکوژنومیکس همچنان پدیدار و رشد می یابند، نهاد های نظارتی بین
المللی دستورالعملهای فارماکوژنومیکس و مقررات را توسعه میدهند. با این حال،
افزایش توجه و پتانسیل مستند و تعهد استراتژیهای توسعه دارو مبتنی بر فارماکوژنومیکس
، مقاومت مداومی برابر این رویکرد در بخش شرکتهای داروسازی وجود داشته است. دلیل
این مقاومت، این درک است که یک استراتژی فارماکوژنومیکس و تحلیل بیوانفورماتیک
منجر به از بین رفتن قابل توجه ضرر حاصل از تجزیه و تحلیل بازار دارو میشود.
علاوه بر این، چنین ادراکی از بیرون نشاهده میشود و به عنوان “اسطوره” توسط برخی
از مصرف کننده گان در نظر گرفته میشود، آنها گزارش دادند که استراتژی
فارماکوژنومیکس پتانسیل افزایش اندازه بازار دارو را در پیش دارد.
موانع پیشرفت در زمینه توسعه و طراحی دارو
فارماکوژنومیکس
براساس تغییرات ژنومی به طور خاص در کد گذاری یا نزدیک به مناطق کدینگ است. پیش بینی
تغییرات ژنی با ابزار های بیوانفورماتیک که بر واکنش دارو اثر میگذارند بسیار
دشوار است. چند شکلی های تک نوکلیوتیدی نقش مهمی در پاسخ به داروی متغیر ایفا میکند. SNPs ها
هر ۱۰۰ – ۳۰۰
باز در امتداد ژنوم انسانی با سه میلیارد باز اتفاق میافتد؛ بنابراین، میلیون ها
از این ها باید شناسایی و توسط نرم افزار های بیوانفورماتیک آنالیز شوند تا دخالت
آنها در واکنش به دارو تعیین شود. آگاهی محدود و دانش نسبت به رابطه بین انواع ژن
و واکنش متغیر دارو نیز به عنوان عاملی محدود کننده برای طراحی دارو و فرآیند تحویل
دارو عمل میکند . از آنجا که بسیاری از ژنها به احتمال زیاد بر پاسخها اثر می
گذارند، به دست آوردن یک تصویر بزرگ از اثر تغییرات ژن به شدت وقت گیر و پیچیده
است، و برای این منظور، ما به پروفایل ژنتیکی هر فرد نیاز داریم که در آیندهای
نزدیک امکان پذیر نیست و حتی با نرم افزار های بیوانفورماتیکی در حال حاظر بسیاز
زمان بر می باشد . پزشکان همچنین باید یک مرحله تشخیصی اضافی را اجرا کنند تا مشخص
شود کدام دارو برای هر بیمار مناسب است. برای تفسیر دقیق تشخیصی و توصیه بهترین
دوره درمان برای هر بیمار، تمام پزشکان تجویز بدون توجه به تخصص به درکی بهتر از
ژنتیک نیاز خواهند داشت. برخی ملاحظات اخلاقی نیز باید پیش از اجرای بالینی روتین
فارماکوژنومیکس انجام شوند. در عین حال، اقتصاد آزمایش فارماکوژنومیکس از دیدگاه بیماران،
پزشکان، شرکتهای بیمه، دولتها، و شرکتهای داروسازی نقش مهمی در تعیین استفاده
از آینده آن ایفا خواهد کرد.
نتیجهگیری
طراحی دارو یک فرآیند
بسیار پیچیده، پرهزینه و زمانبر است. هم بیوانفورماتیک و هم فارماکوژنومیکس پشتیبانی
زیادی را برای غلبه بر هزینه و شرایط زمان فراهم میکنند. بیوانفورماتیک توصیف
کننده طیف گستردهای از پایگاه های داده و نرم افزارهای مرتبط با دارو است که میتواند
برای اهداف مختلف، مربوط به طراحی و فرآیند توسعه دارو مورد استفاده قرار گیرد.
همچنین، فارماکوژنومیکس اطلاعات سطح ژنوم را در مورد واکنش دارو متغیر فراهم می
کند، که برای شرکت های داروسازی بسیار مهم است که داروهای جدید طراحی کنند اگرچه،
بیوانفورماتیک و فارماکوژنومیکس هنوز در مرحله اولیه خود هستند و در حال حاضر با
موانعی مواجه هستند که به اندازه کافی پتانسیل لازم برای کمک به روند توسعه دارو
در آینده نزدیک را نشان میدهند.
خالها رشد بیش از حد سلولهایی از پوست به نام ملانوسیت هستند. هرچند خالها، مانند تومورها رشد بیش از حد سلولها محسوب میشوند، تقریبا اغلب غیرسرطان زا (خوش خیم) هستند. تعداد مشابه خال در افراد نسلهای مختلف یک خانواده، نشان می دهد که تمایل به ایجاد خال ارثی است، اما الگوی وراثت آن کاملا مشخص نشده است. گاهی خالها در زمان تولد یا در طی نوزادی وجود دارند. این خالها که خال مادرزادی نامیده می شوند، تقریبا همواره خوش خیم هستند. به ندرت یک خال بسیار بزرگ که خال ملانوسیتی مادرزادی غول پیکر نامیده می شود، در زمان تولد وجود دارد. در موارد نادر، مهمترین نوع سرطان پوست (به نام ملانوما) در این نوع خال ایجاد میشود. خالهای رنگی، بزرگ و شکل نامنظم را خالهای دیس پلاستیک یا خالهای آتیپیک مینامند که در هر سنی ایجاد میشود. هرچند رایج نیست اما آنها تمایل دارند که زیاد شوند. این خال ها ریسک ابتلا به ملانوما را افزایش میدهند. وراثت در تکوین خالهای دیس پلاستیک و داشتن تعداد بیش از میانگینِ خالهای خوش خیم نقش دارد. سپری کردن زمان زیادی زیر خورشید میتواند تعداد خالهای یک فرد را افزایش دهد. اما خالها بسیاری اوقات در نواحی از بدن که در معرض مواجهه با نور خورشید نبوده اند نیز یافت میشود که نشان دهنده این است فاکتورهای دیگر علاوه بر نور فرابنفش خورشید، مانند هورمونها یا سایر فرایندهای بیولوژیک، در شروع تشکیل خال ملانوسیتی اکتسابی و خال دیس پلاستیک دست دارد. تنوع در چندین ژن از جمله FGFR3, PIK3CA, HRAS و BRAF به خالهای خوش خیم مربوط میشوند. جهش در BRAF منجر به تولید یک پروتئین تغییر یافته می شود که سبب تجمع ملانوسیتها در خال میشود. این پروتئین تغییر یافته همچنین آغاز گر تولید پروتئین سرکوب کنندهی تومور به نام p15 است که رشد بیش از حد خال را متوقف میکند تا خیلی بزرگ نشود. در موارد نادر، جهشهای BRAF همراه با حذف ژن CDKN2A سبب کمبود p15 شده، که باعث می شود رشد خال از کنترل خارج و خال سرطانی شود (بدخیم). وقتی این قضیه با فاکتورهای محیطی نظیر آسیب سلولی ناشی از مواجهه با تشعشع فرابنفش ترکیب شود احتمال ایجاد سرطان بالاتر میرود. درافرادی که پوست روشن، موهای بور دارند، خانواده ای که سابقهی ملانوما دارد، یا در افرادی که ریسک فاکتورهای ژنتیکی نظیر جهش در ژن CDKN2A وجود دارد، تشعشع فرابنفش خورشید میتواند به خالهای فرد آسیب بزند و ریسک بدخیم شدن خال ها را افزایش دهد. تحقیقات نشان داده است افرادی که خالهای زیادی دارند ریسک بالاتری برای ابتلا به ملانوما دارند. اما همچنین افراد مبتلا به ملانومایی تشخیص داده شده اند که خالهای کمی داشته اند و ملانوما در نواحی از بدن که در معرض نور خورشید نبوده است ایجاد شده است. محققان برای برای درک بهتر ژنتیک خالها و رابطه آنها با سرطان در حال تلاشند.
سرطان ریه بیماری است که مشخصه آن رشد کنترلنشده سلولها در بافت ریه است. اگر این بیماری درمان نشود، رشد سلولی میتواند در یک فرایند به نام متاستاز به بیرون از ریه گسترش پیدا کند و به بافتهای اطراف یا سایر اعضای بدن برسد. اکثر سرطانهایی که از ریه شروع میشوند، به نام سرطانهای ابتدایی ریه، کارسینوماهایی هستند که از بافت پوششی نشات میگیرند و در اثر تغییر در ژنهای مشخصی در بدن ایجاد میشوند. در برخی از موارد، این جهشهای ژنی ممکن است از یکی از افراد خانواده که به این بیماری مبتلا است، به ارث برسد. هرچند در بیشتر بیماران مبتلا به سرطان ریه، جهش ژنتیکی در اثر مواجهه با عوامل سرطان زای محیطی، ایجاد می شود. دو مورد رایج از ژنهایی که جهش در آنها با سرطان ریه مرتبط است، شامل EGFR و KRAS هستند. شایعترین علت سرطان ریه قرار گرفتن در معرض دود دخانیات برای یک مدت طولانی است که دلیل نود درصد از سرطانهای ریه است. درصد ابتلا به سرطان ریه در افرادی که سیگار نمیکشند پانزده درصد است، و دلیل این موارد اغلب ترکیبی از عوامل ژنتیکی، گاز رادون، آزبست، و آلودگی هوا از جمله دود سیگار فرد ثالث مربوط میشود. تشخیص سرطان ریه با یک بافتبرداری که اغلب از طریق برونکوسکوپی یا راهنمای سیتی انجام میشود، قابل تایید شدن است. درمان و نتایج طولانی مدت به نوع سرطان، مرحله (شدت گسترش) و سلامت کلی فرد، که از طریق وضعیت عملکرد سنجیده میشود، بستگی دارد. درمانهای رایج عبارتند از جراحی، شیمیدرمانی، و پرتودرمانی. اپی ژنتیک نقش مهمی در پیشرفت و توسعه سرطان ریه دارد. مطالعات نشان داده است که متیلاسیون ژنهای TSG با پیش آگهی سلولهای NSCLC در مراحل اولیه ارتباط دارد. اپی ژنوم ممکن است در درمان سرطان ریه موثر باشد زیرا می تواند مسیرهای متعددی که ویژگیهای اصلی سلولهای سرطانی را تنظیم می کنند، هدف قرار دهد. علیرغم برخی نتایج بالینی ناامید کننده، که فقط درمان اپی ژنتیکی انجام شده بود، این زمینه از علم همچنان در حال تکامل است.
بیماری هموکروماتوز یک بیماری ژنتیکی است، در این بیماری آهن بیش از حد معمول از طریق مواد غذایی در بدن جذب می شود که منجر به غلظت بیش از حد آهن در خون می شود، در این بیماری در تولید یکی از پروتئینهای بدن جهش رخ داده است. این پروتئین وظیفه انتقال آهن را در سطح روده بر عهده دارد که موجب انتقال آهن به داخل پرزهای روده میشود، به دلیل جهش در این پروتئین، عملکرد این پروتئین مختل شده و جذب آهن تا چندین برابر در بدن افزایش مییابد. هموکروماتوز به دو نوع اولیه و ثانویه دسته بندی می شود که نوع اولیه ارثی (ناشی از توارث ژن HFF) و نوع ثانویه در شرایط خاص مانند: کم خونی، بیماریهای کبدی، گرفتن خون بسیار زیاد و… در فرد بروز می نماید. مردان مبتلا به این بیماری 5 برابر بیشتر از زنان می باشد. علائم بیماری در صورت بروز عبارتند از: ضعف، دیابت، درد شکم، خستگی، درد مفاصل، نارسایی کبد، نارسایی قلبی، ناتوانی جنسی. افراد با دارای آهن غیرطبیعی باید تحت انجام تست های ژنتیکی برای تایید تشخیص این بیماری قرار بگیرند. بررسی ژنتیکی این بیماری در آژمایشگاه ژنتیک نسل سالم انجام می شود.
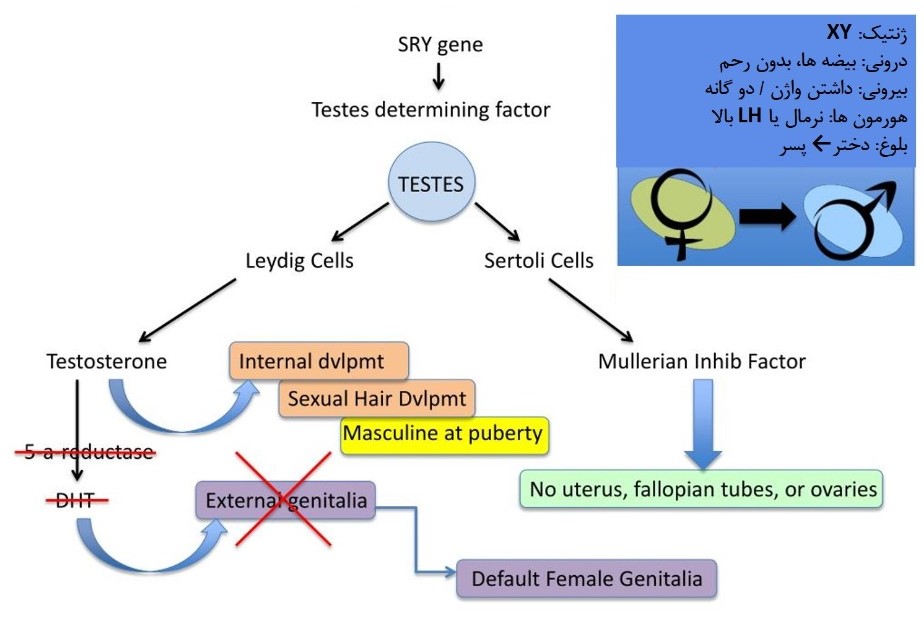
نقص پنج آلفا ردوکتاز یک بیماری ژنتیکی با الگوی توارث اتوزومی مغلوب است که تکوین جنسی افراد مذکر را پیش از تولد و طی بلوغ تحت تاثیر قرار می دهد. افراد مبتلا به این بیماری از نظر ژنتیکی مذکر هستند، دارای یک کروموزوم X و یک کروموزوم Y در هر سلول می باشند، و دارای گنادهای مذکر (بیضه ها) می باشند. بدن آن ها یک هورمون به نام دی هیدرو تستسترون (DHT) را به میزان کافی تولید نمی کند. DHT دارای نقشی ضروری در تکوین جنسی افراد مذکر می باشد و کمبود این هورمون تشکیل پیش از تولد اندام های جنسی خارجی را مختل می کند. بیشتر افراد مبتلا در داشتن فرزند بیولوژیک بدون استفاده از روش های کمک باروری ناتوان هستند. کودکان مبتلا به نقص پنج آلفا ردوکتاز اغلب به عنوان دختر پرورش می یابند. برخی از این افراد در نوجوانی یا اوایل بزرگسالی نقش جنس مرد و افراد دیگر نقش جنس زن را اقتباس می کنند. بررسی ژنتیکی این بیماری در آزمایشگاه ژنتیک نسل سالم قابل انجام می باشد.
فیبروزکیستیک یک اختلال ژنتیک از نوع اتوزوم مغلوب است. این بیماری به دلیل اختلال در عملکرد کانال پروتئینی موجود در سطح سلول ها که مسئول انتقال یون کلر هستند به وجود می آید که در اثر اختلال در عملکرد این کانال ها، انتقال یونهای کلر با بار منفی دچار اختلال شده و به دنبال آن نقل و انتقال آب نیز در بافت های مخاطی بدن دچار مشکل می شود. نتیجه این اختلال چسبناک شدن و غلیظ شدن همه ی مخاط ها بوده که در نهایت می تواند منجر به اختلال در عملکرد ریه ها (عفونت های مکرر ریوی)، انسداد سیستم گوارش و حتی ناباروری شود، با این حال بیشتر مرگ و میر ناشی از فیبروزکیستیک در اثر درگیری ریه میباشد. درمان فیبروزکیستیک بر مبنای بهداشت تهاجمی راه هوایی، حمایت تغذیهای شامل جایگزینی آنزیمهای پانکراس، آنتیبیوتیک ها و گشاد کنندههای مجراهای هوایی می باشد، آنزیم DNAase انسانی نوترکیب استنشاقی (درناز آلفا dornase alfa) چسبندگی خلط را کاهش داده، عملکرد ریهها را بهبود بخشیده، و از تشدید بیماری میکاهد. محلول نمکی هیپرتونیک استنشاقی به آبدار کردن ترشحات کمک کرده و امکان میدهد تا به سادگی با سرفه خارج شوند، و نیز عملکرد ریوی را بهبود میبخشد. تأیید و تشخیص این بیماری در شیرخوارگی و یا اوایل کودکی براساس نتایج تست عرق انجام میشود. در این آزمایش مقدار کلر موجود در عرق کودک اندازهگیری می شود، اگر میزان کلر موجود در عرق کودک مبتلا از فرد معمولی بالاتر باشد با توجه به نتایج تست عرق و علایم بالینی بیمار، تشخیص تأئید می شود. این بیماری در اثر جهش در ژن CFTR به وجود می آید که در آزمایشگاه ژنتیک نسل سالم جهش های مربوط به این ژن مورد بررسی قرار میگیرد. مشاوره ژنتیک برای ناقلین CF قبل یا بعد از ازدواج خیلی ضروری است. همچنین مشاوره ژنتیک برای تمامی خانواده هایی که در فامیل آنها حداقل یک فرد مبتلا به این بیماری وجود دارد (یا قبلا وجود داشته) توصیه می شود.
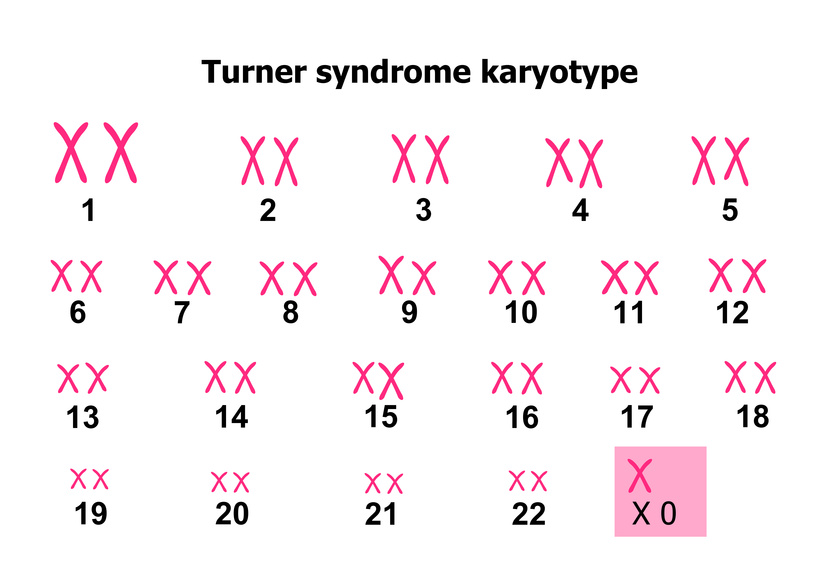
سندروم ترنر جزء آنیوپلوئیدیهای جنسی است، در سندروم ترنر، نوزاد دختر به جای داشتن دو کروموزوم X، تنها یک کرومووزم X دارد، سندروم ترنر بعد از سندروم داون شایعترین اختلال کروموزومی در انسان است. نوزادان دچار سندروم ترنر در معرض خطر بالایی برای مرگ در دوران نوزادی قرار دارند، بطوریکه ۹۸٪ از تمام جنینهای مبتلا به سندروم ترنر به طور خودبخودی سقط میشوند. در صورت زنده ماندن زنان مبتلا اغلب کوتاه قد و نازا بوده، آمنوره اولیه دارند. سندروم ترنر تنها مونوزومی سازگار با طبیعت است که فرصت تولد پیدا میکند. با انجام تست غربالگری سل فری (NIPT) در آزمایشگاه ژنتیک نسل سالم می توان بیماری سندروم ترنر را در جنین از هفته 10 بارداری با دقت و حساسیت بالا تشخیص داد.